Feeling overwhelmed by complex medical terms and unclear information about mitochondrial disease? You’re not alone. This guide breaks down the science behind mitochondrial disorders—from DNA mutations to energy production challenges—into clear, actionable insights for families and patients. Discover how cutting-edge research and patient-centered strategies are shaping better understanding and management of this complex condition.
Table of contents
- Understanding Mitochondrial Disease: Basics and Definitions
- Genetic Origins and Inheritance Patterns
- Types of Mitochondrial Disease and Their Symptoms
- Diagnosis, Treatment and Disease Management
Understanding Mitochondrial Disease: Basics and Definitions
What Are Mitochondria and Their Function
Mitochondria are tiny organelles in cells responsible for energy production. They convert nutrients into adenosine triphosphate (ATP), the body’s energy currency. This process, called oxidative phosphorylation, fuels important functions like muscle contractions and nerve signaling. Proper mitochondrial function is crucial for high-energy tissues like the brain and heart. The Liam Foundation emphasizes their role in cellular health.
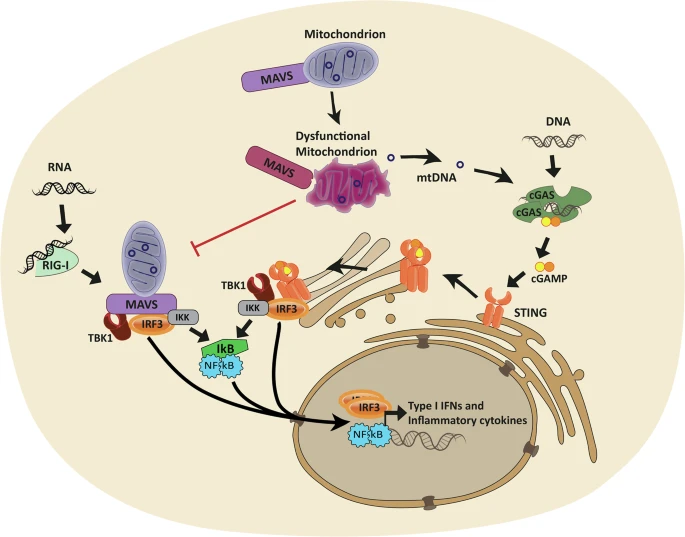
Mitochondria generate 90% of cellular energy through a complex process involving the electron transport chain and ATP synthesis. They rely on both mitochondrial DNA (mtDNA) and nuclear DNA for proper function. Mutations disrupt energy production, impacting organs like muscles, the brain, and the liver. This disruption underlies mitochondrial diseases, which affect energy-dependent tissues first. Mitochondria also regulate apoptosis and calcium balance, highlighting their broader cellular importance.
Defining Mitochondrial Disease and Disorders
Mitochondrial disease encompasses genetic disorders caused by mtDNA or nuclear DNA mutations. These defects impair energy production, leading to cellular dysfunction. Over 300 forms exist, affecting 1 in 4,000 births. Symptoms vary based on which tissues—like muscles, the brain, or heart—experience energy deficits. Genetic testing helps identify specific mutations causing these disorders.
| Category | Primary Mitochondrial Disorders | Secondary Mitochondrial Dysfunction |
|---|
| Origin | Genetic mutations (mtDNA or nuclear DNA) | Environmental factors, toxins, or secondary conditions |
| Energy Production | Direct impairment of ATP synthesis | Indirect effects on mitochondrial function |
| Examples | MELAS, MERRF, Leigh syndrome | Drug-induced (e.g., HIV treatments), aging-related decline |
Mitochondrial dysfunction most severely impacts energy-hungry organs like the brain, muscles, and heart. This explains symptoms such as muscle weakness, neurological issues, and cardiac complications. The variability in symptom presentation reflects differences in mutation types and tissue involvement. As outlined in Nature Reviews Endocrinology, clinical manifestations depend on mitochondrial systems fail and the severity of energy deficits.
Genetic Origins and Inheritance Patterns
Mitochondrial disease arises from mutations in mitochondrial DNA (mtDNA) or nuclear DNA. mtDNA mutations, inherited maternally, affect mitochondrial function directly. Nuclear DNA mutations involve genes like POLG and TPRMB, which regulate mitochondrial processes. These defects disrupt energy production, causing diverse symptoms. The Liam Foundation explains how genetic factors shape disease presentation.
- Mutations in mtDNA cause mitochondrial disease
- mtDNA deletions disrupt mitochondrial energy production
- mtDNA depletion syndromes caused by nuclear gene mutations (TK2, POLG, DGUOK, SUCLA2) reduce mitochondrial DNA levels
- impair mitochondrial replication and repair
mtDNA is passed exclusively from mothers, explaining maternal inheritance patterns. Nuclear DNA mutations follow autosomal dominant or recessive patterns. For example, MELAS syndrome follows maternal inheritance, while POLG-related disorders follow autosomal patterns. These variations impact family planning, as risks differ between mutation types. Ontario data shows 1 in 3,989 people hospitalized with mitochondrial disease between 1988–2019, highlighting genetic transmission’s role in prevalence.
Spontaneous mutations account for cases without family history. These de novo changes occur during early development or gamete formation. Global estimates suggest 1 in 4,000 births involve mitochondrial disorders. While most follow inheritance patterns, new mutations contribute to disease emergence, explaining why some patients lack familial connections to mitochondrial disease.
Types of Mitochondrial Disease and Their Symptoms
Common Mitochondrial Disease Syndromes
Mitochondrial disease includes syndromes like MELAS and MERRF, caused by genetic mutations affecting energy production. These disorders impact high-energy tissues such as the brain and muscles. Symptoms vary widely, from neurological issues to muscle weakness. Over 300 forms exist, with MELAS and MERRF being among the most studied. Early diagnosis helps tailor care for better outcomes.
Detailed Description of MELAS Syndrome
MELAS involves stroke-like episodes, lactic acidosis, and muscle weakness. It’s linked to mtDNA mutations, notably m.3243A>G in MT-TL1. Patients often experience seizures and migraines. Lactic acid buildup causes metabolic stress. Brain imaging shows lesions outside typical stroke regions. Symptoms emerge in childhood, progressing unpredictably. Genetic testing confirms diagnosis. Treatments focus on symptom management and metabolic support, though no cure exists.
Explanation of MERRF Syndrome
MERRF features myoclonus epilepsy and ragged red fibers in muscle biopsies. Caused by MTTK mutations (A8344G in 80% of cases), it disrupts mitochondrial protein synthesis. Patients may develop ataxia and hearing loss. Onset typically occurs in childhood or adolescence. The condition’s hallmark is clusters of abnormal mitochondria in muscle cells, visible under specialized staining.
Less Common Mitochondrial Disorders
Leigh syndrome affects infants with neurological decline and respiratory issues. NARP (neuropathy, ataxia, retinitis pigmentosa) stems from MT-ATP6 mutations, causing vision loss and coordination problems. These rarer forms highlight mitochondrial DNA’s role in diverse organ systems. Their clinical diversity complicates diagnosis but underscores the need for comprehensive genetic analysis.
Detailed Description of Leber’s Hereditary Optic Neuropathy and NARP
Leber’s hereditary optic neuropathy (LHON) causes sudden vision loss due to mtDNA mutations (MT-ND1, MT-ND4, MT-ND6). NARP involves retinal degeneration, peripheral neuropathy, and ataxia from MT-ATP6 variants. Both disorders demonstrate tissue-specific mitochondrial dysfunction. LHON primarily affects young adults, while NARP presents with progressive neurological decline. Early genetic testing is crucial for management.
Explanation of Kearns-Sayre Syndrome and DNA Depletion Syndromes
Kearns-Sayre syndrome features progressive external ophthalmoplegia, retinal degeneration, and heart block. It’s tied to mtDNA deletions, often sporadic. Mitochondrial DNA depletion syndromes (MTDPS) result from nuclear gene mutations (TK2, POLG), reducing mtDNA levels. These syndromes cause severe myopathy or liver failure. Their autosomal recessive inheritance means both parents carry the mutation.
Symptoms Across Body Systems
Organ-Specific Symptoms of Mitochondrial Disease| Organ/System | Common Symptoms | Additional Notes |
|---|
| Heart | Cardiomyopathy, arrhythmias | Requires regular cardiac monitoring; can lead to heart failure |
| Brain | Stroke-like episodes, seizures, developmental delays | Characteristic of MELAS; linked to brain lesions |
| Muscles | Weakness, fatigue, exercise intolerance | Reduced stamina and pain during activity |
| Eyes | Vision loss (LHON), ptosis, retinitis pigmentosa | LHON causes sudden vision loss; NARP includes retinal degeneration |
| Nervous System | Ataxia, neuropathy | Impacts coordination and nerve function |
| Digestive System | Motility issues (vomiting, diarrhea, constipation) | Swallowing or nutrient absorption challenges |
| Liver | Acute failure, elevated enzymes | Frequent in pediatric cases |
| Kidneys | Tubular dysfunction, electrolyte imbalances | Impairs waste filtration and fluid balance |
| Ears | Sensorineural hearing loss | Progressive loss from cochlear nerve damage |
| Pancreas | Diabetes mellitus | Beta-cell dysfunction |
Symptom Variability and Progression
Symptoms vary due to mutation type, tissue energy demands, and heteroplasmy levels. Some experience stable symptoms; others face progressive decline. Environmental factors and secondary conditions can trigger fluctuations. Energy deficits often worsen over time without intervention. Regular monitoring helps adapt care as disease evolves.
Common Pathogenic Mechanisms
Energy failure underlies all mitochondrial disorders, but high-energy tissues like the brain and muscles show earliest damage. mtDNA mutations accumulate in postmitotic cells, while nuclear DNA defects disrupt mitochondrial replication. This dual genetic origin explains diverse clinical presentations. Addressing energy deficits remains central to therapeutic strategies.
Diagnosis, Treatment and Disease Management
Diagnostic Approaches and Challenges
Mitochondrial disease diagnosis involves clinical evaluation, family history analysis, and specialized testing. Doctors assess symptoms across organs like the brain, muscles, and heart. Bloodwork, genetic testing (mtDNA and nuclear DNA), and muscle biopsies help confirm diagnosis. Globally, 1 in 4,000 births is affected, with Ontario recording 3,069 hospitalizations between 1988–2019. Family history is important due to maternal or autosomal inheritance patterns. Genetic testing identifies mutations like POLG or MELAS, while imaging and biomarkers support clinical findings.
- Blood and urine tests for lactic acidosis and metabolic markers
- Imaging (MRI, CT) for brain or organ involvement
- Muscle biopsy for ragged red fibers and enzyme activity
- Genetic testing (mtDNA sequencing, nuclear gene panels)
- Biomarkers like elevated CSF lactate and plasma pyruvate
Genetic testing clarifies mitochondrial disease through mtDNA and nuclear DNA analysis. Whole-exome sequencing identifies pathogenic variants in genes like POLG. ClinGen’s Mitochondrial Diseases Gene Curation Expert Panel validates over 250 nuclear genes linked to mitochondrial disorders. Next-generation sequencing improves variant detection accuracy. Prenatal testing and carrier screening guide family planning. Emerging technologies like single-cell sequencing may refine diagnosis further, addressing current limitations in identifying low-level heteroplasmy.
Treatment Options and Management Strategies
Current mitochondrial disease management focuses on symptom relief and supportive care. No cure exists, so treatment personalizes to organ involvement. Supplements like CoQ10 and L-carnitine address energy production deficits. Dietary strategies (high-fat, low-carb) help some patients. Physical therapy maintains mobility, while medications target seizures or cardiac issues. Regular monitoring prevents complications. Support programs guide families through daily challenges.
Therapies include CoQ10 for mitochondrial dysfunction and antioxidant support. Creatine monohydrate improves energy storage in muscles. Carnitine aids fatty acid transport into mitochondria. Ketogenic or modified Atkin’s diet benefits certain epilepsy-related syndromes. Avoid valproate due to liver toxicity risks. For MELAS, dichloroacetate may reduce lactic acidosis. Cardiac issues require pacemakers. Vision loss in LHON needs neuroprotective agents. Multidisciplinary care coordinates interventions across neurology, cardiology, and nutrition.
Patients benefit from controlled exercise programs to boost mitochondrial biogenesis. Low-intensity aerobic activity (e.g., walking) improves stamina without overexertion. Energy conservation techniques involve scheduling rest breaks and pacing activities. Environmental factors like extreme heat or cold worsen symptoms, so temperature regulation is important. Adaptive equipment (e.g., mobility aids) eases daily tasks. Sleep hygiene supports energy recovery. Work closely with specialists to balance activity and recovery.
Emerging Treatments and Research Directions
Research explores gene therapy for mtDNA mutations and nucleoside supplementation to stabilize mitochondrial DNA. EPI-743, a synthetic CoQ10 analog, shows promise in clinical trials. Antioxidants like idebenone improve vision in LHON. Cutting-edge research includes mitochondrial replacement therapy and small molecules targeting the electron transport chain. Stem cell approaches aim to repair defective mitochondria in affected tissues. These innovations may one day address root causes, but current focus remains on quality-of-life improvements through symptom management.
Mitochondrial disease stems from genetic mutations disrupting cellular energy, impacting vital organs like the brain and muscles. Early diagnosis through genetic testing and personalized management—such as supplements or lifestyle adjustments—can stabilize symptoms. As research advances, staying informed and proactive offers families hope for improved therapies and a future where mitochondrial dysfunction is met with clearer answers and stronger support.
Questions & Answer about Mitochondrial Disease